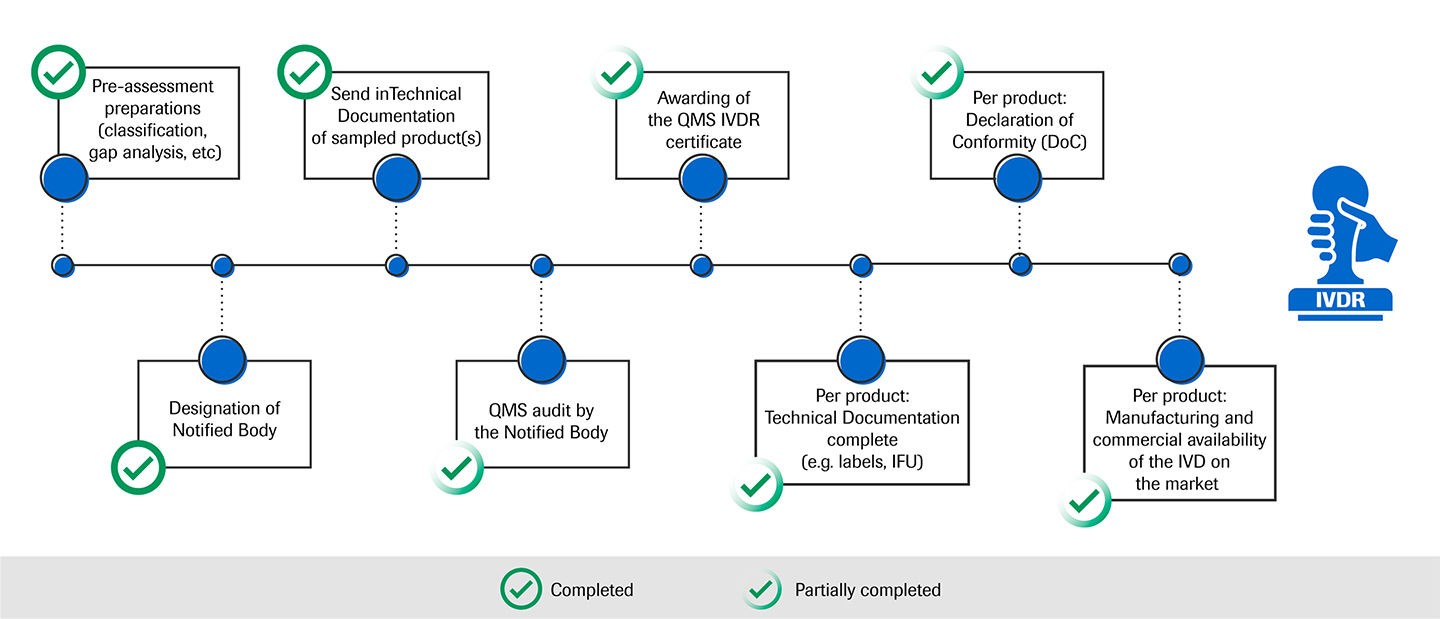
Il 17 dicembre Roche ha annunciato di aver ricevuto il primo "Certificato del sistema di gestione qualità per l'UE (IVDR)" da TÜV SÜD Product Service, uno dei propri enti notificati ai sensi del nuovo Regolamento per i dispositivi medico-diagnostici in vitro (IVDR). La certificazione si applica a più di 700 prodotti del portfolio Area Siero e Coagulazione di laboratorio.
Il presente "Certificato del sistema di gestione qualità per l'UE (IVDR)" è obbligatorio per tutti i produttori di dispositivi diagnostici in vitro (IVD) affinché possano emettere dichiarazioni di conformità per i rispettivi prodotti e ottenerne la (ri)certificazione in conformità al nuovo regolamento.
Il nostro percorso verso la conformità IVDR
Ci impegniamo a gestire l’importante transizione dalla Direttiva sulla diagnostica in vitro (IVDD) al Regolamento sulla diagnostica in vitro (IVDR) attivamente e con tempestività. La nostra priorità è soddisfare i requisiti normativi e le esigenze dei nostri clienti per garantire la disponibilità dei nostri prodotti necessari agli operatori sanitari e ai pazienti. Per raggiungere la piena conformità IVDR, stiamo seguendo un approccio in più fasi: